

Over the years, our group has identified many previously uncharacterized receptors and binding proteins, notably TNFR1 & 2 (with Prof. D. Wallach), IFNAR2, and IL-18BP (with Prof. C. Dinarello). In 1993, we found that a naturally occurring soluble form of the LDL receptor (LDLR) inhibits infection of cells by vesicular stomatitis virus (VSV). Because LDLR-deficient cells were still infected by VSV we could not determine if LDLR is a VSV receptor (Fischer, D.G. et al, Science, 262:250-3). We now report that LDLR is the major VSV receptor, but VSV can infect cells also through other LDLR family members (PNAS online Apr. 15th, 2013).
Why is VSV an important virus?
VSV exhibits a remarkably robust and pantropic infectivity, mediated by its coat protein, VSV-G. Utilizing this property, recombinant forms of VSV and VSV-G-pseudotyped viral vectors are being developed for gene therapy, vaccination and viral oncolysis, and are extensively employed for laboratory gene transduction in vivo and in vitro.
What did we find?
The broad tropism of VSV suggests that it enters cells through a highly ubiquitous receptor, whose identity has so far remained elusive. Using a combination of monoclonal antibody against LDLR and recombinant Receptor Associated Protein (RAP), we completely blocked VSV entry and infectivity (Fig. 1). Addition of RAP to the culture medium completely blocked transduction of LDLR-deficient cells by VSV-G-pseudotyped lentiviral vector (Fig. 2). These results indicate that LDLR serves as the major entry port of VSV and of VSV-G-pseudotyped lentiviral vectors in human and mouse cells, whereas other LDLR family members serve as alternative receptors.
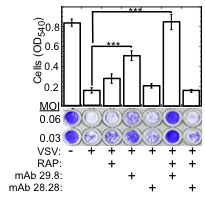
Fig. 1. LDLR and its family members serve as VSV receptors.
Crystal violet stained WISH cells grown to confluence in 96 well plates, incubated (30 min., 37°C) with the indicated combinations of RAP (200 nM), neutralizing anti LDLR mAb 29.8, and non-neutralizing anti LDLR mAb 28.28 (50 µg/ml each); cells were then challenged with VSV at the indicated MOI. Cell viability (bar plot) was determined by reading the OD540 of cultures treated with VSV at MOI=0.06. ***P<0.002, n=4.
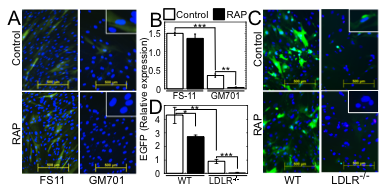
Fig. 2. LDLR and its other family members serve as major and minor VSV entry ports in human and mouse cells.
(A) EGFP expression in WT human FS-11 fibroblasts and LDLR-deficient GM701 fibroblasts, transduced with EGFP-encoding VSV-G-pseudotyped lentiviral vector (VSV-G-LV) in the absence (Control) or presence of RAP (100 nM). Insets: higher magnifications. Bars=500 µm. (B) Average±SD of EGFP expression shown in panel A. ***P<0.0002, n=3. *P<0.03, n=3. (C) EGFP expression in WT murine embryonic fibroblasts (WT MEFs) and LDLR-deficient MEFs, transduced with EGFP-encoding VSV-G-LV as in A. Insets: higher magnifications. Bars=500 µm. (D) Average±SD of EGFP expression shown in panel C. All fluorescence intensity values were normalized to the nuclei counts. *P<0.05, **P<0.007, ***P<0.002, n=3.
Why are these findings remarkable?
The widespread expression of LDLR family members accounts for the pantropism of VSV, and for the broad applicability of VSV-G-pseudotyped viral vectors for gene transduction. Our study closes a quest for the VSV receptor that started in 1983, when phosphatidylserine was proposed as a possible VSV receptor and later shown not to be the VSV receptor.
The identification of the VSV receptor is of significant clinical importance since recombinant VSV and VSV-G-pseudotyped viral vectors are being developed for viral oncolysis, for vaccination and for gene therapy. The recently develped CAR-T therapy makes use of VSV-G- coated viral vectors for construction of the T cells. Upregulation of LDLR in vivo, e.g., by pre-treatment with statins might increase the efficacy of such vectors. Furthermore, liver cells and certain tumor cells, which express high levels of LDLR, might be the preferred targets of VSV-G based gene therapy as well as VSV-G-based viral oncolysis.