IceQream (IQ) is an interpretable and robust modeling framework that predicts chromatin accessibility changes directly from DNA sequence using mechanistic models of transcription factor (TF) binding. Unlike black-box deep learning approaches, IQ achieves competitive—or superior—performance while maintaining full biological transparency.
IQ models TF-sequence recognition by spatial integration of sequences over a range of affinities and localization relative to the target locus. It quantifies TF effective concentrations as latent variables that activate or repress regulatory elements in a non-linear fashion, with contributions from pairwise interactions and synergistic chromosomal domain effects.
IQ’s white-box design allows inference and synthesis of models explaining accessibility dynamics over an entire single-cell manifold, providing insights into the regulatory logic controlling cell state transitions.
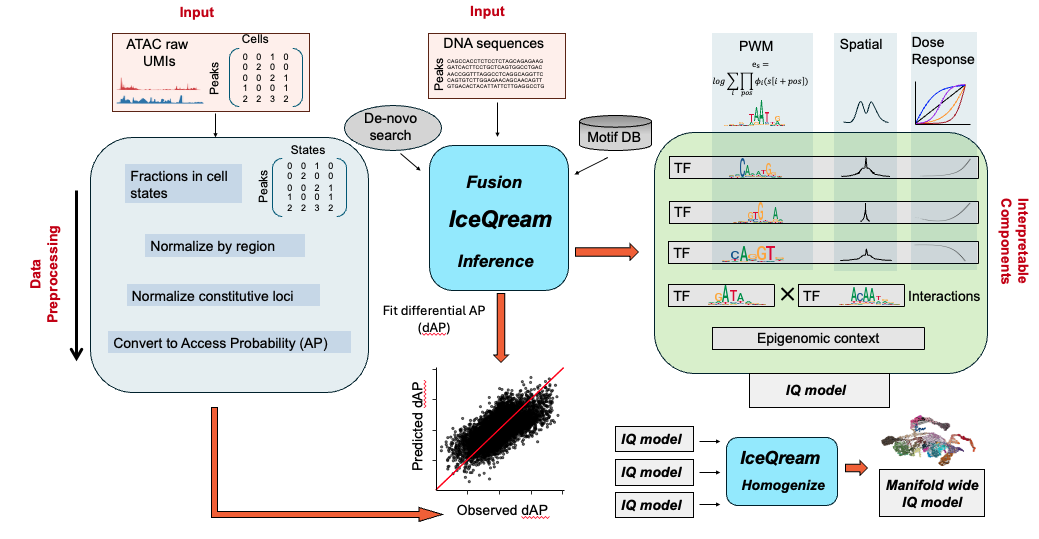
Schematic of the IceQream (IQ) workflow: (i) Single-cell ATAC raw counts are transformed into estimated access probabilities (AP). (ii) The IQ model incorporates transcription factor (TF) models, epigenomic context variables, and pairwise interactions of TF models. Each TF model integrates contributions from strong and weak affinity sequences, weighted by spatial preferences around the accessible hotspot, which are transformed into dose-response-like spatial binding preferences curves using pre-defined non-linear functions. (iii) Model initialization involves scanning candidate TF models from PSSM (position-specific scoring matrix) databases and de-novo motif regression. (iv) An integrated IQ model predicts differential AP (dAP) across a selected manifold trajectory. (v) IQ models from multiple trajectories are fused to create a manifold-wide set of common TF motif models.