Our group combines mathematical modelling, large-scale biomedical data and experiments, to understand human physiology, aging and disease. We are physicists, biologists, computer scientists and MDs working together to form the basic equations of hormone circuits, antibiotics, autoimmunity, cancer, mood disorders and age-related diseases. Our style emphasizes teaching good communication skills, listening and having fun being creative while going together into the unknown.
Pregnancy and delivery involve dynamic alterations in many physiological systems. However, the physiological dynamics during pregnancy and after delivery have not been systematically analyzed at high temporal resolution in a large human population. Here, we present the dynamics of 76 lab tests based on a cross-sectional analysis of 44 million measurements from over 300,000 pregnancies. We analyzed each test at weekly intervals from 20 weeks preconception to 80 weeks postpartum, providing detailed temporal profiles. About half of the tests take 3 months to a year to return to baseline postpartum, highlighting the physiological load of childbirth. The precision of the data revealed effects of preconception supplements, overshoots after delivery and intricate temporal responses to changes in blood volume and renal filtration rate. Pregnancy complicationsgestational diabetes, preeclampsia, and postpartum hemorrhageshowed distinct dynamical changes. These results provide a comprehensive dynamic portrait of the systems physiology of pregnancy.
Miyara S., Adler M., Umansky K. B., Häußler D., Bassat E., Divinsky Y., Elkahal J., Kain D., Lendengolts D., Ramirez Flores R. O., Bueno-Levy H., Golani O., Shalit T., Gershovits M., Weizman E., Genzelinakh A., Kimchi D. M., Shakked A., Zhang L., Wang J., Baehr A., Petrover Z., Sarig R., Dorn T., Moretti A., Saez-Rodriguez J., Kupatt C., Tanaka E. M., Medzhitov R., Krüger A., Mayo A., Alon U. & Tzahor E.
(2025)
Cell Systems.
16,
3,
101198.
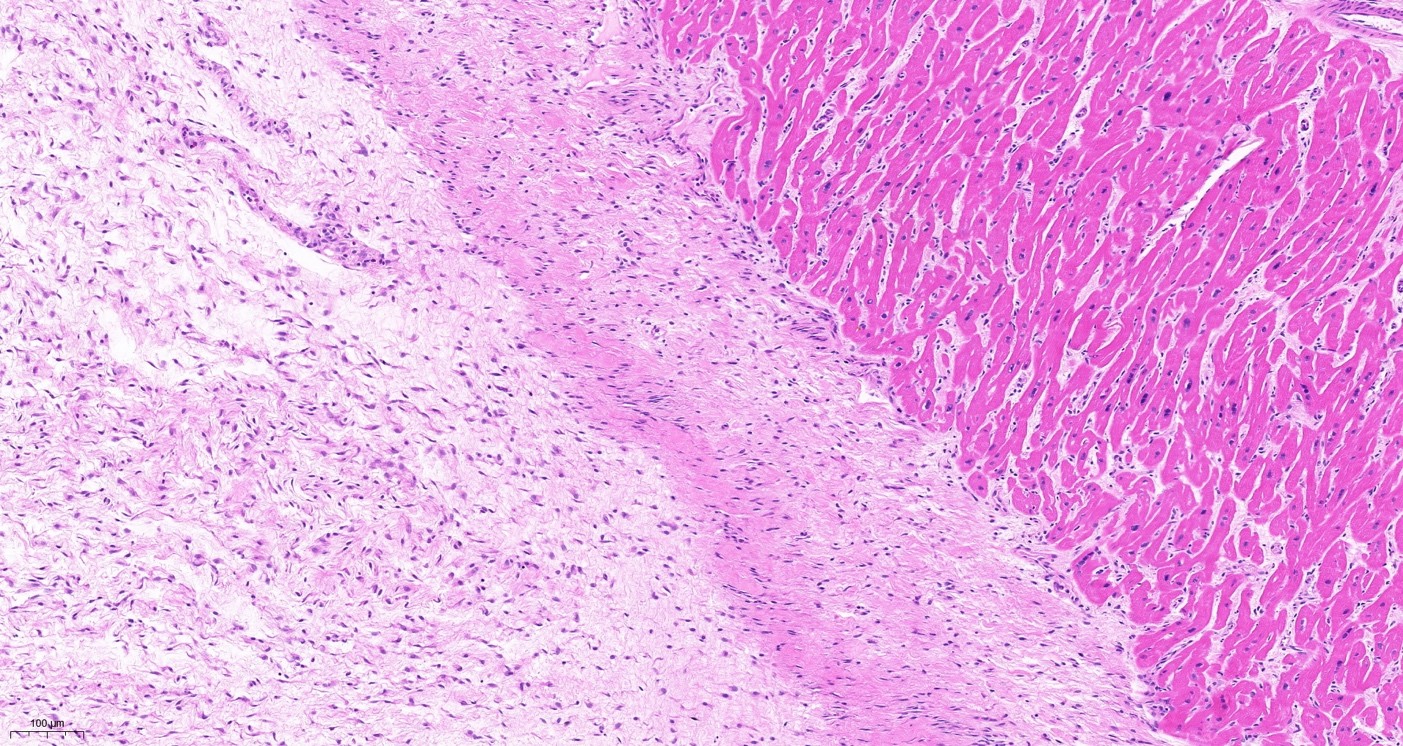
Fibrosis remains a major unmet medical need. Simplifying principles are needed to better understand fibrosis and to yield new therapeutic approaches. Fibrosis is driven by myofibroblasts that interact with macrophages. A mathematical cell-circuit model predicts two types of fibrosis: hot fibrosis driven by macrophages and myofibroblasts and cold fibrosis driven by myofibroblasts alone. Testing these concepts in cardiac fibrosis resulting from myocardial infarction (MI) and heart failure (HF), we revealed that acute MI leads to cold fibrosis whereas chronic injury (HF) leads to hot fibrosis. MI-driven cold fibrosis is conserved in pigs and humans. We computationally identified a vulnerability of cold fibrosis: the myofibroblast autocrine growth factor loop. Inhibiting this loop by targeting TIMP1 with neutralizing antibodies reduced myofibroblast proliferation and fibrosis post-MI in mice. Our study demonstrates the utility of the concepts of hot and cold fibrosis and the feasibility of a circuit-to-target approach to pinpoint a treatment strategy that reduces fibrosis. A record of this paper's transparent peer review process is included in the supplemental information.
Yang Y., Karin O., Mayo A., Song X., Chen P., Santos A. L., Lindner A. B. & Alon U.
(2023)
Nature Communications.
14,
2209.
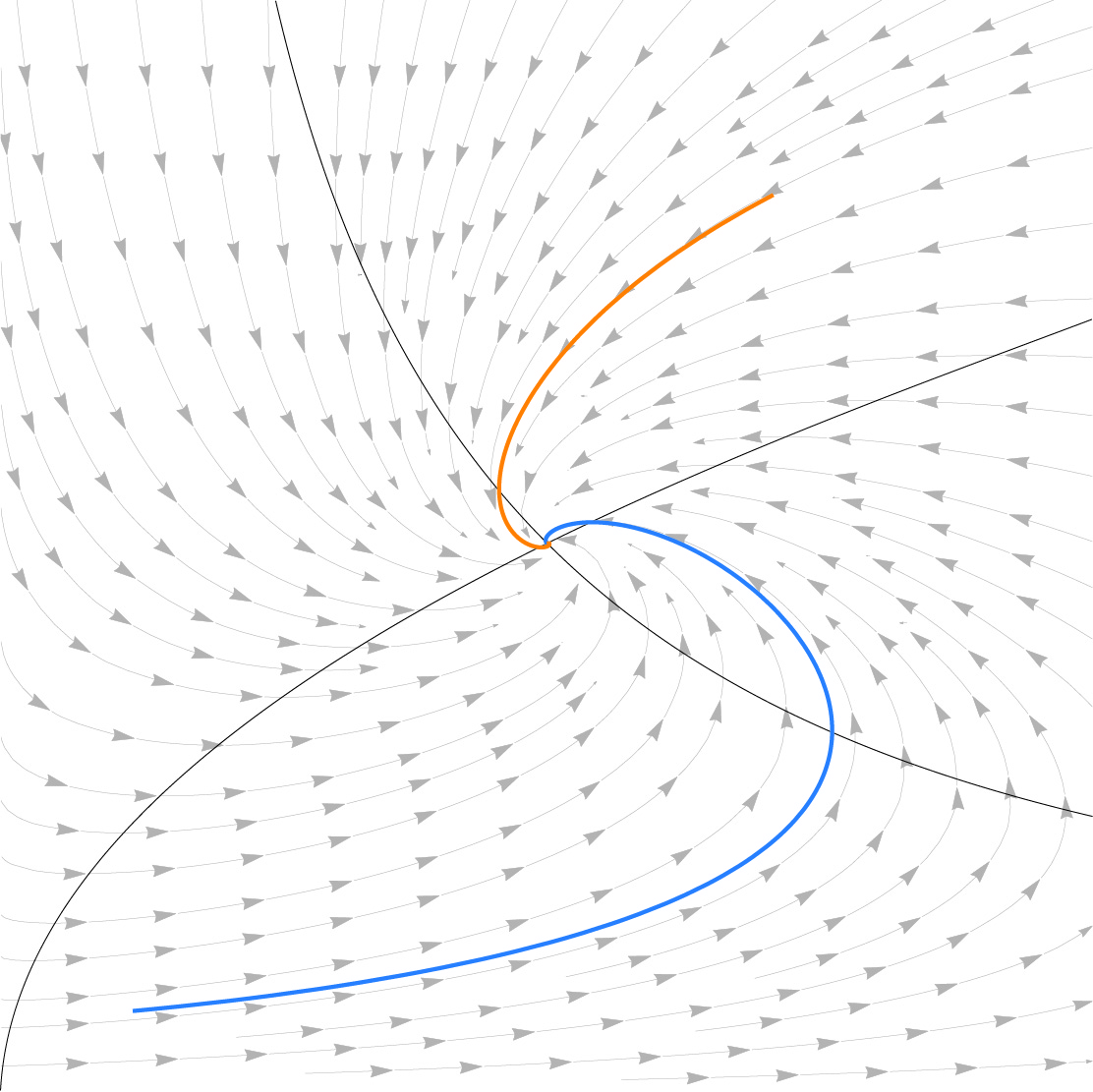
Genetically identical cells in the same stressful condition die at different times. The origin of this stochasticity is unclear; it may arise from different initial conditions that affect the time of demise, or from a stochastic damage accumulation mechanism that erases the initial conditions and instead amplifies noise to generate different lifespans. To address this requires measuring damage dynamics in individual cells over the lifespan, but this has rarely been achieved. Here, we used a microfluidic device to measure membrane damage in 635 carbon-starved Escherichia coli cells at high temporal resolution. We find that initial conditions of damage, size or cell-cycle phase do not explain most of the lifespan variation. Instead, the data points to a stochastic mechanism in which noise is amplified by a rising production of damage that saturates its own removal. Surprisingly, the relative variation in damage drops with age: cells become more similar to each other in terms of relative damage, indicating increasing determinism with age. Thus, chance erases initial conditions and then gives way to increasingly deterministic dynamics that dominate the lifespan distribution.